Coronaviruses
Please note that this page is no longer updated. ECDC is currently developing a factsheet for health professionals on COVID-19 which will be made available in the first quarter of 2023.
This section is aimed at assisting public health professionals and is based on an ongoing rapid review of the latest evidence.
(Last update 15 August 2022)
General background
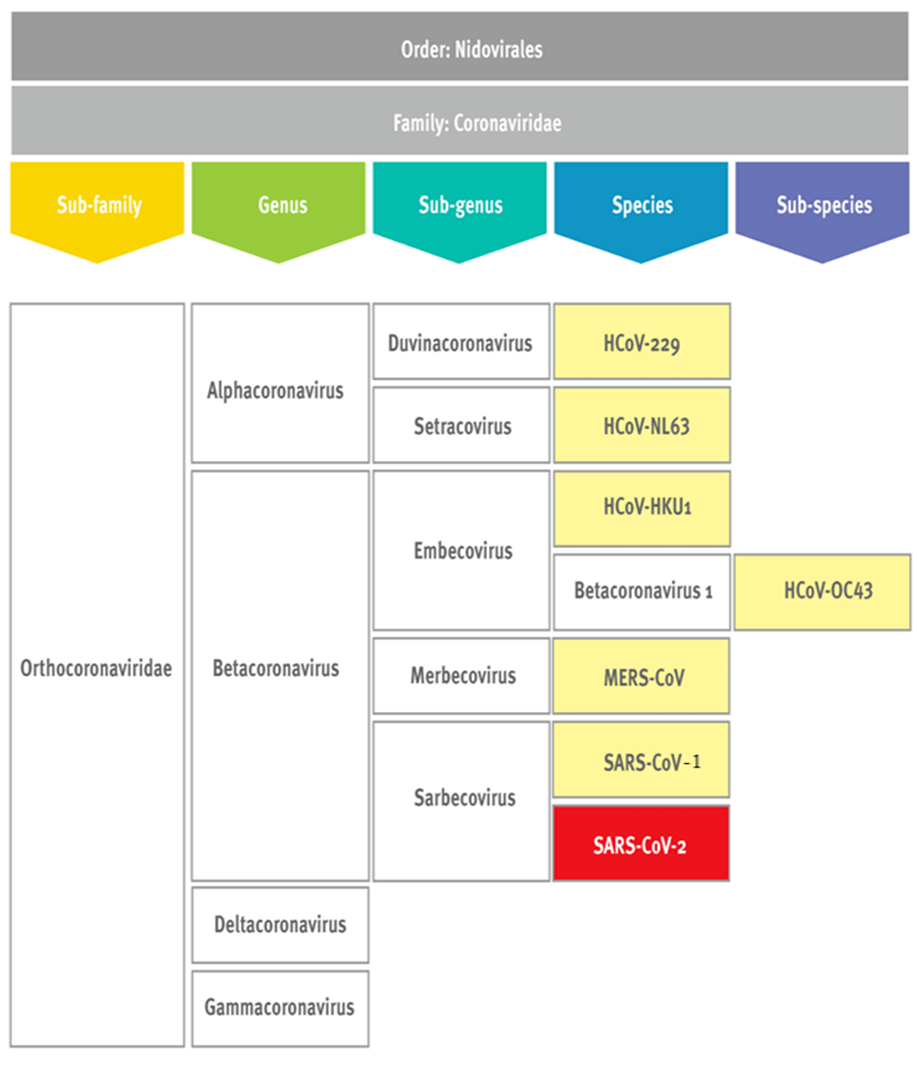
Coronaviruses (CoVs) were identified as human pathogens in the 1960s. They are enveloped, positive-stranded RNA viruses in the order of Nidovirales (Figure) [1]. The characteristic surface of a coronavirus virion has a crown-like appearance that can be seen under the electron microscope, which is why the viruses are named after the Latin word corona, meaning crown or halo.
Most coronaviruses infect animals (i.e. bats, birds and mammals), which act as reservoirs and intermediate hosts, but can sometimes change host and infect humans.
There are currently seven coronaviruses known to infect humans (Figure). Four of them cause mild-to-moderate disease: HCoV-OC43, HCoV-HKU1 and HCoV-229E cause common colds (but may cause severe lower respiratory tract infections among people in the youngest and oldest age groups), while HCoV-NL63 is an important cause of (pseudo) croup and bronchiolitis in children [2]. The other three cause more severe and possibly even fatal disease and have emerged more recently: SARS-CoV-1, responsible for severe acute respiratory syndrome (SARS), which emerged in 2002; MERS-CoV, responsible for Middle East respiratory syndrome (MERS), which emerged in 2012; and SARS-CoV-2, responsible for coronavirus disease 2019 (COVID-19), which was identified in late 2019 [3,4].
Illness in humans mostly affects the respiratory tract, with symptoms ranging from those of a common cold to very severe lower respiratory infection [5].
Figure: Human coronavirus taxonomy
SARS-CoV-2 virus evolution
The GISAID EpiCoV database holds SARS-CoV-2 genome sequences. Some caution needs to be taken when interpreting the results in the database due to variations in reporting and sampling timeframes between countries.
The current hypothesis is that the virus emerged in or close to the city of Wuhan in Hubei province, China, where it was first detected. However, so far, no direct ancestor to the virus has been found that can fully explain its emergence [6]. Studies using SARS-CoV-2 genome data suggest that the first human cases of the pandemic likely emerged shortly before (earliest estimate: October 2019) confirmed cases were detected in Hubei province in December 2019 [7,8]. The scientific evidence supports a zoonotic origin of SARS-CoV-2 [9].
An analysis of genome sequence data suggests that SARS-CoV-2 was introduced from Hubei province into EU/EEA countries on different occasions and that the predominant lineage from the early phases of the virus’s spread has a most recent common ancestor in Italy [10]. The earliest detection of the virus in the EU/EEA comes from retrospective analysis of sewage samples in Milan and Turin, which showed that the virus was present in northern Italy on 18 December 2019 [11].
Three different time-calibrated phylogenetic analyses of closely related coronaviruses suggest that the lineage giving rise to SARS-CoV-2 diverged from the most similar known bat coronavirus between 1948 and 1982 [12]. Bats are the most likely original animal reservoir of the virus, with an intermediate animal host probably involved in the transmission to humans, as has been the case for other coronaviruses [12,13]. However, the recombinant nature of coronaviruses complicates longer-term phylogenetic analysis, making it difficult to disentangle the ancestry of SARS-CoV-2. Genomic evidence also indicates that the virus is unlikely to be a product of in-vitro manipulation or passaging in cell culture, or of synthetic origin [6,9,14].
As for all viruses, SARS-CoV-2 will constantly change through mutation. Indeed, many variants of SARS-CoV-2 with different sets of mutations have been observed worldwide. While most emerging SARS-CoV-2 variants will not have a significant impact on the spread of the virus, some mutations or combinations of mutations may provide the virus with a selective advantage, such as increased transmissibility or the ability to evade the host immune response. These variants could increase the risk posed by SARS-CoV-2 to human health, which is why they are considered to be variants of concern (VOCs).
Some variants have emerged that represent significant evolutionary jumps, with no intermediate forms detected. These variants include all the VOCs classified so far. The three main hypotheses presented for these jumps are: i) prolonged infections, likely in immunocompromised patients, which could have allowed the virus to accumulate mutations to avoid the host immune response without any of the evolutionary bottlenecks associated with human-to-human transmission; ii) transmission of SARS-CoV-2 to an animal host (most likely wild) and prolonged circulation in an animal population, where mutations could then have accumulated; and iii) circulation in countries with little or no genomic surveillance, where mutations could have accumulated without being detected. Of the three hypotheses, the prolonged infections hypothesis has the most direct supporting evidence, with studies showing that immune escape mutations can arise during prolonged infections, particularly in combination with treatment with convalescent plasma [15]. SARS-CoV-2 can infect many animal species, and some accumulation of mutations associated with circulation among animals has been demonstrated [16]. Due to screening of travellers and extensive genomic surveillance programs in many countries, it is unlikely that a variant could emerge through undetected circulation and gradual accumulation of mutations. More direct evidence is needed in order to determine which hypothesis or combination of hypotheses best explains the evolutionary jumps associated with some SARS-CoV-2 variants.
ECDC is continuously monitoring the emergence and circulation of SARS-COV-2 VOCs in the EU/EEA. The following resources can be consulted for more information:
- Details on emerging VOCs and VOCs circulating in the EU/EEA can be found in ECDC risk assessments.
- A map of the distribution of current VOCs based on sequenced samples from the EU/EEA is available from the GISAID EpiCoV database.
- A map showing the weekly sequencing volumes by country, using data submitted to the European Surveillance system (TESSy) and the GISAID EpiCoV database, is available in ECDC’s weekly COVID-19 country overview.
- General information about SARS-CoV-2 VOCs, variants of interest (VOIs) and variants under monitoring can be found on ECDC’s website.
References
- International Committee on Taxonomy of Viruses (ICTV). Virus Taxonomy: The Classification and Nomenclature of Viruses. The 9th Report of the ICTV. ICTV; 2011. Available at: https://talk.ictvonline.org/ictv-reports/ictv_9th_report/
- Yin Y, Wunderink RG. MERS, SARS and other coronaviruses as causes of pneumonia. Respirology. 2018 Feb;23(2):130-7. Available at: https://www.ncbi.nlm.nih.gov/pubmed/29052924
- World Health Organization (WHO). WHO Statement regarding cluster of pneumonia cases in Wuhan, China. Geneva: WHO; 2020. Available at: https://www.who.int/china/news/detail/09-01-2020-who-statement-regarding-cluster-of-pneumonia-cases-in-wuhan-china
- World Health Organization (WHO). Disease outbreak news update. Novel Coronavirus – China 2020. Geneva: WHO; 2020. Available at: https://www.who.int/emergencies/disease-outbreak-news/item/2020-DON233
- Channappanavar R, Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin Immunopathol. 2017 Jul;39(5):529-39. Available at: https://www.ncbi.nlm.nih.gov/pubmed/28466096
- Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS-CoV-2. Nat Med. 2020 Apr;26(4):450-2. Available at: https://www.ncbi.nlm.nih.gov/pubmed/32284615
- Pekar J, Worobey M, Moshiri N, Scheffler K, Wertheim JO. Timing the SARS-CoV-2 index case in Hubei province. Science. 2021 Apr 23;372(6540):412-7. Available at: https://www.ncbi.nlm.nih.gov/pubmed/33737402
- van Dorp L, Acman M, Richard D, Shaw LP, Ford CE, Ormond L, et al. Emergence of genomic diversity and recurrent mutations in SARS-CoV-2. Infect Genet Evol. 2020 Sep;83:104351. Available at: https://www.ncbi.nlm.nih.gov/pubmed/32387564
- Holmes EC, Goldstein SA, Rasmussen AL, Robertson DL, Crits-Christoph A, Wertheim JO, et al. The origins of SARS-CoV-2: A critical review. Cell. 2021 Sep 16;184(19):4848-56. Available at: https://www.ncbi.nlm.nih.gov/pubmed/34480864
- Nadeau SA, Vaughan TG, Scire J, Huisman JS, Stadler T. The origin and early spread of SARS-CoV-2 in Europe. Proc Natl Acad Sci USA. 2021 Mar 2;118(9). Available at: https://www.ncbi.nlm.nih.gov/pubmed/33571105
- La Rosa G, Mancini P, Bonanno Ferraro G, Veneri C, Iaconelli M, Bonadonna L, et al. SARS-CoV-2 has been circulating in northern Italy since December 2019: Evidence from environmental monitoring. Sci Total Environ. 2021 Jan 1;750:141711. Available at: https://www.ncbi.nlm.nih.gov/pubmed/32835962
- Boni MF, Lemey P, Jiang X, Lam TT, Perry BW, Castoe TA, et al. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat Microbiol. 2020 Nov;5(11):1408-17. Available at: https://www.ncbi.nlm.nih.gov/pubmed/32724171
- Hu B, Zeng LP, Yang XL, Ge XY, Zhang W, Li B, et al. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 2017 Nov;13(11):e1006698. Available at: https://www.ncbi.nlm.nih.gov/pubmed/29190287
- Othman H, Bouslama Z, Brandenburg JT, da Rocha J, Hamdi Y, Ghedira K, et al. Interaction of the spike protein RBD from SARS-CoV-2 with ACE2: Similarity with SARS-CoV, hot-spot analysis and effect of the receptor polymorphism. Biochem Biophys Res Commun. 2020 Jun 30;527(3):702-8. Available at: https://www.ncbi.nlm.nih.gov/pubmed/32410735
- Corey L, Beyrer C, Cohen MS, Michael NL, Bedford T, Rolland M. SARS-CoV-2 Variants in Patients with Immunosuppression. N Engl J Med. 2021 Aug 5;385(6):562-6. Available at: https://www.ncbi.nlm.nih.gov/pubmed/34347959
- Zhou P, Shi ZL. SARS-CoV-2 spillover events. Science. 2021 Jan 8;371(6525):120-2. Available at: https://www.ncbi.nlm.nih.gov/pubmed/33414206